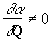| |||||||||||
|
Raman Technical
Resources
See other sections of KOSI's website for information on:
1. A brief look at Raman scattering theory. 1.1. The Raman Effect and Normal Raman Scattering. When light is scattered from a molecule most photons are elastically scattered. The scattered photons have the same energy (frequency) and, therefore, wavelength, as the incident photons. However, a small fraction of light (approximately 1 in 107 photons) is scattered at optical frequencies different from, and usually lower than, the frequency of the incident photons. The process leading to this inelastic scatter is the termed the Raman effect. Raman scattering can occur with a change in vibrational, rotational or electronic energy of a molecule. Chemists are concerned primarily with the vibrational Raman effect. We will use the term Raman effect to mean vibrational Raman effect only. The difference in energy between the incident photon and the Raman scattered photon is equal to the energy of a vibration of the scattering molecule. A plot of intensity of scattered light versus energy difference is a Raman spectrum. 1.1.1. The Scattering Process. The Raman effect arises when a photon is incident on a molecule and interacts with the electric dipole of the molecule. It is a form of electronic (more accurately, vibronic) spectroscopy, although the spectrum contains vibrational frequencies. In classical terms, the interaction can be viewed as a perturbation of the molecule’s electric field. In quantum mechanics the scattering is described as an excitation to a virtual state lower in energy than a real electronic transition with nearly coincident de-excitation and a change in vibrational energy. The scattering event occurs in 10-14 seconds or less. The virtual state description of scattering is shown in Figure 1.1a.
The energy difference between the incident and scattered photons is
represented by the arrows of different lengths in Figure 1.1a.
Numerically, the energy difference between the initial and final
vibrational levels,
in which l incident and l scattered are the wavelengths (in cm) of the incident and Raman scattered photons, respectively. The vibrational energy is ultimately dissipated as heat. Because of the low intensity of Raman scattering, the heat dissipation does not cause a measurable temperature rise in a material. At room temperature the thermal population of vibrational excited states is low, although not zero. Therefore, the initial state is the ground state, and the scattered photon will have lower energy (longer wavelength) than the exciting photon. This Stokes shifted scatter is what is usually observed in Raman spectroscopy. Figure 1.1a depicts Raman Stokes scattering. A small fraction of the molecules are in vibrationally excited states. Raman scattering from vibrationally excited molecules leaves the molecule in the ground state. The scattered photon appears at higher energy, as shown in Figure 1.1b. This anti-Stokes-shifted Raman spectrum is always weaker than the Stokes-shifted spectrum, but at room temperature it is strong enough to be useful for vibrational frequencies less than about 1500 cm-1. The Stokes and anti-Stokes spectra contain the same frequency information. The ratio of anti-Stokes to Stokes intensity at any vibrational frequency is a measure of temperature. Anti-Stokes Raman scattering is used for contactless thermometry. The anti-Stokes spectrum is also used when the Stokes spectrum is not directly observable, for example because of poor detector response or spectrograph efficiency. 1.1.2. Vibrational Energies. The energy of a vibrational mode depends on molecular structure and environment. Atomic mass, bond order, molecular substituents, molecular geometry and hydrogen bonding all effect the vibrational force constant which, in turn dictates the vibrational energy. For example, the stretching frequency of a phosphorus-phosphorus bond ranges from 460 to 610 to 775 cm-1 for the single, double and triple bonded moieties, respectively.[1] Much effort has been devoted to estimation or measurement of force constants. For small molecules, and even for some extended structures such as peptides, reasonably accurate calculations of vibrational frequencies are possible with commercially available software. Vibrational Raman spectroscopy is not limited to intramolecular vibrations. Crystal lattice vibrations and other motions of extended solids are Raman-active. Their spectra are important in such fields as polymers and semiconductors. In the gas phase, rotational structure is resolvable on vibrational transitions. The resulting vibration/rotation spectra are widely used to study combustion and gas phase reactions generally. Vibrational Raman spectroscopy in this broad sense is an extraordinarily versatile probe into a wide range of phenomena ranging across disciplines from physical biochemistry to materials science. 1.1.3. Raman Selection Rules and Intensities. A simple classical electromagnetic field description of Raman spectroscopy can be used to explain many of the important features of Raman band intensities. The dipole moment, P, induced in a molecule by an external electric field, E, is proportional to the field as shown in equation 2.
The proportionality constant a is the polarizability of the molecule. The polarizability measures the ease with which the electron cloud around a molecule can be distorted. The induced dipole emits or scatters light at the optical frequency of the incident light wave. Raman scattering occurs because a molecular vibration can change the
polarizability. The change is described by the polarizability
derivative,
The Raman selection rule is analogous to the more familiar selection rule for an infrared-active vibration, which states that there must be a net change in permanent dipole moment during the vibration. From group theory it is straightforward to show that if a molecule has a center of symmetry, vibrations which are Raman-active will be silent in the infrared, and vice versa. Scattering intensity is proportional to the square of the induced
dipole moment, that is to the square of the polarizability
derivative, If a vibration does not greatly change the polarizability, then the polarizability derivative will be near zero, and the intensity of the Raman band will be low. The vibrations of a highly polar moiety, such as the O-H bond, are usually weak. An external electric field can not induce a large change in the dipole moment and stretching or bending the bond does not change this. Typical strong Raman scatterers are moieties with distributed electron clouds, such as carbon-carbon double bonds. The pi-electron cloud of the double bond is easily distorted in an external electric field. Bending or stretching the bond changes the distribution of electron density substantially, and causes a large change in induced dipole moment. Chemists generally prefer a quantum-mechanical approach to Raman scattering theory, which relates scattering frequencies and intensities to vibrational and electronic energy states of the molecule. The standard perturbation theory treatment assumes that the frequency of the incident light is low compared to the frequency of the first electronic excited state. The small changes in the ground state wave function are described in terms of the sum of all possible excited vibronic states of the molecule. 1.1.4. Polarization Effects. Raman scatter is partially polarized, even for molecules in a gas or liquid, where the individual molecules are randomly oriented. The effect is most easily seen with an exciting source which is plane polarized. In isotropic media polarization arises because the induced electric dipole has components which vary spatially with respect to the coordinates of the molecule. Raman scatter from totally symmetric vibrations will be strongly polarized parallel to the plane of polarization of the incident light. The scattered intensity from non-totally symmetric vibrations is 3/4 as strong in the plane perpendicular to the plane of polarization of the incident light as in the plane parallel to it. The situation is more complicated in a crystalline material. In that case the orientation the crystal is fixed in the optical system. The polarization components depend on the orientation of the crystal axes with respect to the plane of polarization of the input light, as well as on the relative polarization of the input and the observing polarizer. 1.2. Resonance-Enhanced Raman Scattering. Raman spectroscopy is conventionally performed with green, red or near-infrared lasers. The wavelengths are below the first electronic transitions of most molecules, as assumed by scattering theory. The situation changes if the wavelength of the exciting laser within the electronic spectrum of a molecule. In that case the intensity of some Raman-active vibrations increases by a factor of 102-104. This resonance enhancement or resonance Raman effect can be quite useful. Metalloporphyrins, carotenoids and several other classes of biologically important molecules have strongly allowed electronic transitions in the visible. The spectrum of the chromophoric moiety is resonance enhanced and that of the surrounding protein matrix is not. This allows the physical biochemist to probe the chromophoric site (often the active site) without spectral interference from the surrounding protein. Resonance Raman spectroscopy is also a major probe of the chemistry of fullerenes, polydiacetylenes and other "exotic" molecules which strongly absorb in the visible. Although many more molecules absorb in the ultraviolet, the high cost of lasers and optics for this spectral region have limited UV resonance Raman spectroscopy to a small number of specialists. The vibrations whose Raman bands are resonance enhanced fall into two or three general classes. The most common case is Franck-Condon enhancement, in which a component of the normal coordinate of the vibration is in a direction in which the molecule expands during an electronic excitation. The more the molecule expands along this axis when it absorbs light, the larger the enhancement factor. The easily visualized ring breathing (in-plane expansion) modes of porphyrins fall into this class. Vibrations which couple two electronic excited states are also resonance enhanced. This mechanism is called vibronic enhancement. In both cases enhancement factors roughly follow the intensities of the absorption spectrum. The theory of resonance enhancement is beyond the scope of this tutorial. The interested reader is referred to specific reviews.[2] Resonance enhancement does not begin at a sharply defined wavelength. In fact, enhancement of 5X-10X is commonly observed if the exciting laser is even within a few hundred wave numbers below the electronic transition of a molecule. This pre-resonance enhancement can be experimentally useful. 1.3. Surface-Enhanced Raman Scattering. The Raman scattering from a compound (or ion) adsorbed on or even within a few Angstroms of a structured metal surface can be 103-106X greater than in solution. This surface-enhanced Raman scattering is strongest on silver, but is observable on gold and copper as well. At practical excitation wavelengths, enhancement on other metals is unimportant. Surface-enhanced Raman scattering (SERS) arises from two mechanisms. The first is an enhanced electromagnetic field produced at the surface of the metal. When the wavelength of the incident light is close to the plasma wavelength of the metal, conduction electrons in the metal surface are excited into an extended surface electronic excited state called a surface plasmon resonance. Molecules adsorbed or in close proximity to the surface experience an exceptionally large electromagnetic field. Vibrational modes normal to the surface are most strongly enhanced. The second mode of enhancement is by the formation of a charge-transfer complex between the surface and analyte molecule. The electronic transitions of many charge transfer complexes are in the visible, so that resonance enhancement occurs. Molecules with lone pair electrons or pi clouds show the strongest SERS. The effect was first discovered with pyridine. Other aromatic nitrogen or oxygen containing compounds, such as aromatic amines or phenols, are strongly SERS active. The effect can also been seen with other electron-rich functionalities such as carboxylic acids. The intensity of the surface plasmon resonance is dependent on many factors including the wavelength of the incident light and the morphology of the metal surface. The wavelength should match the plasma wavelength of the metal. This is about 382 nm for a 5 m m silver particle[3], but can be as high as 600 nm for larger ellipsoidal silver particles. The plasma wavelength is to the red of 650 nm for copper and gold[4], the other two metals which show SERS at wavelengths in the 350-1000 nm region. The best morphology for surface plasmon resonance excitation is a small (<100 nm) particle or an atomically rough surface. SERS is used to study monolayers of materials adsorbed on metals, including electrodes. Many formats other than electrodes can be used. The most popular include colloids, metal films on dielectric substrates and, recently, arrays of metal particles bound to metal or dielectric colloids through short linkages. Although SERS allows easy observation of Raman spectra from solution concentrations in the micromolar (1 X 10-6) range, slow adsorption kinetics and competitive adsorption limit its application in analytical chemistry. References Raman Review Newsletter | Application Notes | Technical Notes | Published Articles | Books | |||||||||||
|

 This is a tutorial-style introduction to the basics
of the Raman effect.
This is a tutorial-style introduction to the basics
of the Raman effect.